Tutorial 2: Analyzing G-Tensors¶
This tutorial demonstrates how to use the python API to manipulate, analyze, and plot G-Tensor data.
Prerequisites¶
Before starting this tutorial, ensure you have:
MuTopia package installed
Download the pre-compiled data to the
tutorial_datadirectory
1. The elements of a G-Tensor¶
[1]:
import mutopia.analysis as mu
import mutopia.plot.track_plot as tr
import numpy as np
[2]:
data=mu.gt.load_dataset("tutorial_data/Liver.nc", with_samples=False)
data
[2]:
<xarray.Dataset> Size: 352MB
Dimensions: (configuration: 2, context: 96, locus: 388247,
sample: 185)
Coordinates:
* configuration (configuration) <U12 96B 'C/T-centered' 'A/G...
* context (context) <U7 3kB 'A[C>A]A' ... 'T[T>C]T'
* locus (locus) int64 3MB 0 1 2 ... 388245 388246
* sample (sample) <U36 27kB '0040b1b6-b07a-4b6e-90ef-...
Data variables: (12/25)
Features/GeneExpression (locus) float32 2MB nan nan nan ... nan nan nan
Features/GeneStrand (locus) int8 388kB 0 0 0 0 0 0 ... 0 0 0 0 0 0
Features/GenePosition (locus) float32 2MB nan nan nan ... nan nan nan
Features/ReplicationStrand (locus) int8 388kB 0 0 0 1 1 1 ... 0 0 0 0 0 0
Features/RepliseqS4 (locus) float32 2MB 7.007 2.3 ... 5.495 5.105
Features/NucleotideRatio (locus) float32 2MB 0.3104 0.2271 ... 0.239
... ...
Regions/chrom (locus) <U5 8MB 'chr1' 'chr1' ... 'chr9' 'chr9'
Regions/start (locus) int64 3MB 810000 820000 ... 138190000
Regions/end (locus) int64 3MB 820000 830000 ... 138200000
Regions/length (locus) float32 2MB 3.141e+03 ... 3.408e+03
Regions/context_frequencies (configuration, context, locus) float32 298MB ...
Regions/exposures (locus) float64 3MB 1.0 1.0 1.0 ... 1.0 1.0 1.0
Attributes:
name: liver_simple
dtype: sbs
genome_file: /n/data1/hms/dbmi/park/ctDNA_loci_project/locusregressio...
fasta_file: /n/data1/hms/dbmi/park/SOFTWARE/REFERENCE/hg38/cgap_matc...
blacklist_file: /n/data1/hms/dbmi/park/ctDNA_loci_project/locusregressio...
region_size: 10000
filename: tutorial_data/Liver.nc
regions_file: Liver.nc.regions.bedYou can see the G-Tensor comprises many hierarchical sections with data spanning multiple dimensions. The Dimensions listed at the top indicate the multiple dimensions that may be referenced by the data. In this instance, the G-Tensor contains dimensions for:
locus - regions across the genome
context - the trinucleotide context at which a mutation is found
configuration - configuration indicates whether a mutation was found at a C/T base or an A/G base. Typically, mutational signatures are oriented relative to the C/T base, but we track both orientations to account for stranded features.
sample - the number of samples included in the dataset
You can click on the various “sections” to view their contents. To access variables in a certain section, like “length” under the “Regions” section, you can use:
[3]:
data.sections["Regions"]["length"]
[3]:
<xarray.DataArray 'length' (locus: 388247)> Size: 2MB array([ 3141., 10725., 4845., ..., 6438., 3450., 3408.], dtype=float32) Coordinates: * locus (locus) int64 3MB 0 1 2 3 4 ... 388242 388243 388244 388245 388246
Or:
[4]:
data["Regions/length"]
[4]:
<xarray.DataArray 'Regions/length' (locus: 388247)> Size: 2MB array([ 3141., 10725., 4845., ..., 6438., 3450., 3408.], dtype=float32) Coordinates: * locus (locus) int64 3MB 0 1 2 3 4 ... 388242 388243 388244 388245 388246
In fact the hierarchy is a trick of the UI here. A G-Tensor in fact is simply an X-array dataset where we’ve added a little functionality for variables saved with filepath-like names! I suggest reading a little bit about X-array to understand some of the semantics with dealing in X-array datasets.
Besides the Regions section, which describes the genomic loci, we also have the Features section, which contains our genomic state features, sectioned by cell type (side note: whenever a variable does not belong to a section, it just goes under “Data” - you do not need to prefix with “Data” to access these.)
[5]:
data.sections["Features"]
[5]:
<xarray.Dataset> Size: 35MB
Dimensions: (locus: 388247)
Coordinates:
* locus (locus) int64 3MB 0 1 2 3 ... 388243 388244 388245 388246
Data variables: (12/19)
GeneExpression (locus) float32 2MB nan nan nan nan ... nan nan nan nan
GeneStrand (locus) int8 388kB 0 0 0 0 0 0 0 0 0 ... 0 0 0 0 0 0 0 0
GenePosition (locus) float32 2MB nan nan nan nan ... nan nan nan nan
ReplicationStrand (locus) int8 388kB 0 0 0 1 1 1 1 1 1 ... -1 0 0 0 0 0 0 0
RepliseqS4 (locus) float32 2MB 7.007 2.3 2.088 ... 5.1 5.495 5.105
NucleotideRatio (locus) float32 2MB 0.3104 0.2271 0.1438 ... 0.239 0.239
... ...
H3K4me3 (locus) float32 2MB 0.2934 5.248 0.3616 ... 0.1928 0.1672
RepliseqG1b (locus) float32 2MB 45.48 52.38 52.3 ... 18.0 16.9 17.2
RepliseqS2 (locus) float32 2MB 6.4 6.0 6.1 6.1 ... 29.4 29.7 31.5
RepliseqS1 (locus) float32 2MB 24.92 27.42 27.89 ... 18.4 19.49 21.0
H3K27ac (locus) float32 2MB 0.9603 7.082 ... 0.07612 0.05248
H3K27me3 (locus) float32 2MB 0.0852 0.0844 ... 0.2999 0.1846
Attributes:
name: liver_simple
dtype: sbs
genome_file: /n/data1/hms/dbmi/park/ctDNA_loci_project/locusregressio...
fasta_file: /n/data1/hms/dbmi/park/SOFTWARE/REFERENCE/hg38/cgap_matc...
blacklist_file: /n/data1/hms/dbmi/park/ctDNA_loci_project/locusregressio...
region_size: 10000
filename: tutorial_data/Liver.nc
regions_file: Liver.nc.regions.bedThe methods under mu.gt are operations which operate on G-Tensors. annot_empirical_marginal calculates the average counts for each locus and context across your dataset:
[6]:
data = mu.gt.annot_empirical_marginal(data)
data
Reducing samples: 100%|██████████| 184/184 [00:27<00:00, 6.70it/s]
INFO: Mutopia:Added key: "empirical_marginal"
INFO: Mutopia:Added key: "empirical_marginal_locus"
[6]:
<xarray.Dataset> Size: 652MB
Dimensions: (configuration: 2, context: 96, locus: 388247,
sample: 185)
Coordinates:
* configuration (configuration) <U12 96B 'C/T-centered' 'A/G...
* context (context) <U7 3kB 'A[C>A]A' ... 'T[T>C]T'
* locus (locus) int64 3MB 0 1 2 ... 388245 388246
* sample (sample) <U36 27kB '0040b1b6-b07a-4b6e-90ef-...
Data variables: (12/27)
Features/GeneExpression (locus) float32 2MB nan nan nan ... nan nan nan
Features/GeneStrand (locus) int8 388kB 0 0 0 0 0 0 ... 0 0 0 0 0 0
Features/GenePosition (locus) float32 2MB nan nan nan ... nan nan nan
Features/ReplicationStrand (locus) int8 388kB 0 0 0 1 1 1 ... 0 0 0 0 0 0
Features/RepliseqS4 (locus) float32 2MB 7.007 2.3 ... 5.495 5.105
Features/NucleotideRatio (locus) float32 2MB 0.3104 0.2271 ... 0.239
... ...
Regions/end (locus) int64 3MB 820000 830000 ... 138200000
Regions/length (locus) float32 2MB 3.141e+03 ... 3.408e+03
Regions/context_frequencies (configuration, context, locus) float32 298MB ...
Regions/exposures (locus) float64 3MB 1.0 1.0 1.0 ... 1.0 1.0 1.0
empirical_marginal (configuration, context, locus) float32 298MB ...
empirical_marginal_locus (locus) float32 2MB 0.0001265 3.065e-05 ... 0.0
Attributes:
name: liver_simple
dtype: sbs
genome_file: /n/data1/hms/dbmi/park/ctDNA_loci_project/locusregressio...
fasta_file: /n/data1/hms/dbmi/park/SOFTWARE/REFERENCE/hg38/cgap_matc...
blacklist_file: /n/data1/hms/dbmi/park/ctDNA_loci_project/locusregressio...
region_size: 10000
filename: tutorial_data/Liver.nc
regions_file: Liver.nc.regions.bedYou can see it added keys for empirical_marginal, which has dimensions (context, locus), but also empirical_marginal_locus, which simply sums over the “context” dimension. This is a common pattern in MuTopia methods, giving you access to multiple levels of summarized data.
Another important accessor is mu.gt.fetch_features, which returns a matrix of features which match some query (wildcards with * may be provided):
[7]:
mu.gt.fetch_features(data, "Gene*").to_pandas().T.head(3)
[7]:
| feature | GeneExpression | GeneStrand | GenePosition |
|---|---|---|---|
| locus | |||
| 0 | NaN | 0.0 | NaN |
| 1 | NaN | 0.0 | NaN |
| 2 | NaN | 0.0 | NaN |
2. Manipulating G-Tensors¶
Since G-Tensors are really just souped-up X-arrays, we can do all sorts of fancy manipulations with the data. Mutopia provides a few additional operations like mu.gt.slice_regions and mu.gt.slice_samples.
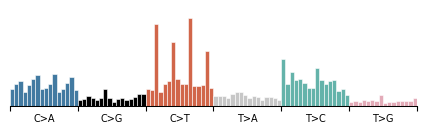
Of particular note is the ability to sum across dimensions. To view the overall length distribution of the data, for example, we can do:
[8]:
data.modality().plot(
data["empirical_marginal"].sum(dim=("locus", "configuration"))
)
[8]:
<Axes: >
One of the unique features of G-Tensors is the ability to load sample data from disk. You can check out the samples your dataset contains with:
[9]:
data.list_samples()[:5]
[9]:
array(['0040b1b6-b07a-4b6e-90ef-133523eaf412',
'00c27940-c623-11e3-bf01-24c6515278c0',
'03c88506-d72e-4a44-a34e-a7f0564f1799',
'0831e45e-c623-11e3-bf01-24c6515278c0',
'0a9c9db0-c623-11e3-bf01-24c6515278c0'], dtype='<U36')
And load the sample data like so:
[10]:
data.fetch_sample("0040b1b6-b07a-4b6e-90ef-133523eaf412")
[10]:
<xarray.Dataset> Size: 2MB
Dimensions: (configuration: 2, context: 96, locus: 388247)
Coordinates:
sample <U36 144B '0040b1b6-b07a-4b6e-90ef-133523eaf412'
Dimensions without coordinates: configuration, context, locus
Data variables:
X (configuration, context, locus) float32 2MB <GCXS: nnz=13687, fill_value=0.0>
ploidy (locus) float64 0B <COO: nnz=0, fill_value=0.0>A sample contains two data elements: the X matrix - the counts of each observed fragment for each fragment length bin at each locus, and ploidy - the copy number at each locus, but transformed such that: p = CN/2 - 1, which allows us to save the data in a sparse manner (since a diploid region corresponds with p=0).
The ability to load data persists through slicing the data:
[11]:
(
mu.gt.slice_regions(data, "chr1:41007569-54616441")
.fetch_sample("0040b1b6-b07a-4b6e-90ef-133523eaf412")
.dims
)
INFO: Mutopia:Found 2343/388247 regions matching query.
[11]:
FrozenMappingWarningOnValuesAccess({'configuration': 2, 'context': 96, 'locus': 2343})
3. Plotting G-Tensor data¶
Plotting G-Tensors works by composing views - sections of the genome, and configs - the configuration of the tracks you want to plot. Configurations themselves are functions of views, and are data-independent descriptions of the plot you want to make.
The general form of a view is a list, tuple, or other iterable of plot elements, like tr.scale_bar, tr.scatterplot, tr.heatmap_plot, etc. Each of these plots are ultimately wrappers around the corresponding matplotlib plot, so you can pass the familiar keyword argument to configure plot aesthetics.
Unlike matplotlib or seaborn, however, you do not directly provide any data to a tr.{plot} element. Instead, the first argument describes how to access that data from a G-Tensor object. For this, you can use the methods:
tr.select- select this variable (or variables) from the G-Tensor.tr.feature_matrix- select the genomic features which match the query string.
Sometimes, you want to plot a transformation of some stored variable. This is simple with tr.pipeline. You simply provide the series of functions you wish to run on the underlying data. For example, to first select some data, then apply the sqrt transformation, one would provide:
tr.pipeline(
tr.select(variable),
lambda x : np.sqrt(x)
)
We provide a few ready-to-go transformations, like and tr.renorm, tr.clip. The tr.apply_rows function applies any provided transformation to each row of a matrix independently. This style of composing configurations enables the construction complex multimodal genome plots. Let’s build one!
(Note: you can specify additional arguments for a config that must be filled at run-time).
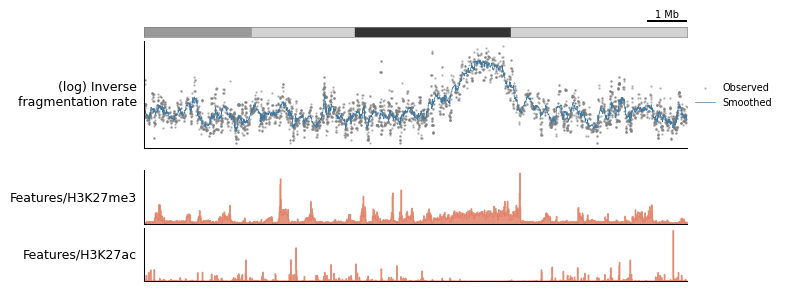
The following example has a little bit of everything:
[12]:
plot_config = lambda view, scalebar_bp : ( #supply scalebar_bp at runtime
tr.scale_bar(scalebar_bp, scale="mb" if scalebar_bp >= 1_000_000 else "kb"),
tr.ideogram("tutorial_data/cytoBand.txt"),
tr.stack_plots( # this context overlays whichever plots are provided
tr.scatterplot(
tr.pipeline(
tr.select("empirical_marginal_locus"),
view.smooth(5),
),
s=0.5,
alpha=0.5,
color="grey",
label="Observed"
),
tr.line_plot(
tr.pipeline(
tr.select("empirical_marginal_locus"),
view.smooth(20),
),
label="Smoothed",
linewidth=0.5,
color=mu.pl.categorical_palette[0], # nice colors
),
height=1.5,
label="(log) Inverse\nfragmentation rate"
),
tr.spacer(0.2),
tr.fill_plot(
tr.select("Features/H3K27me3"),
color=mu.pl.categorical_palette[1],
alpha=0.8,
height=0.75,
),
tr.fill_plot(
tr.select("Features/H3K27ac"),
color=mu.pl.categorical_palette[1],
alpha=0.8,
height=0.75,
)
)
Now, to use the configuration, make a “view” of the data using make_view, supplying the data a region of interest:
[13]:
view = tr.make_view(data, "chr1:41007569-54616441")
INFO: Mutopia:Found 2343/388247 regions matching query.
Finally, use tr.plot_view to render the configuration - make sure to supply any required runtime arguments!
[14]:
tr.plot_view(plot_config, view, scalebar_bp=1_000_000)
None
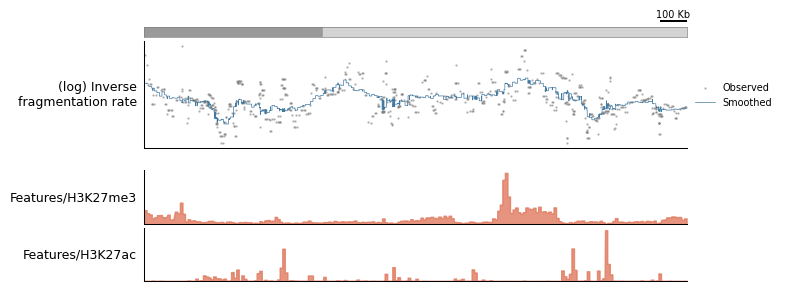
To demonstrate the extensibility of a plotting configuration, let’s say you found an interesting locus and you want to “zoom in”. You can simply re-use the same configuration, modify the run-time paramters, and provide a view of a different region of the genome:
[15]:
zoomed_view = tr.make_view(data, "chr1:43007569-45116441")
tr.plot_view(plot_config, zoomed_view, scalebar_bp=100_000)
None
INFO: Mutopia:Found 403/388247 regions matching query.
Plotting only becomes more important once you’ve trained models on the data - so more on this later!